What are the regulatory requirements for essential documents in clinical research?
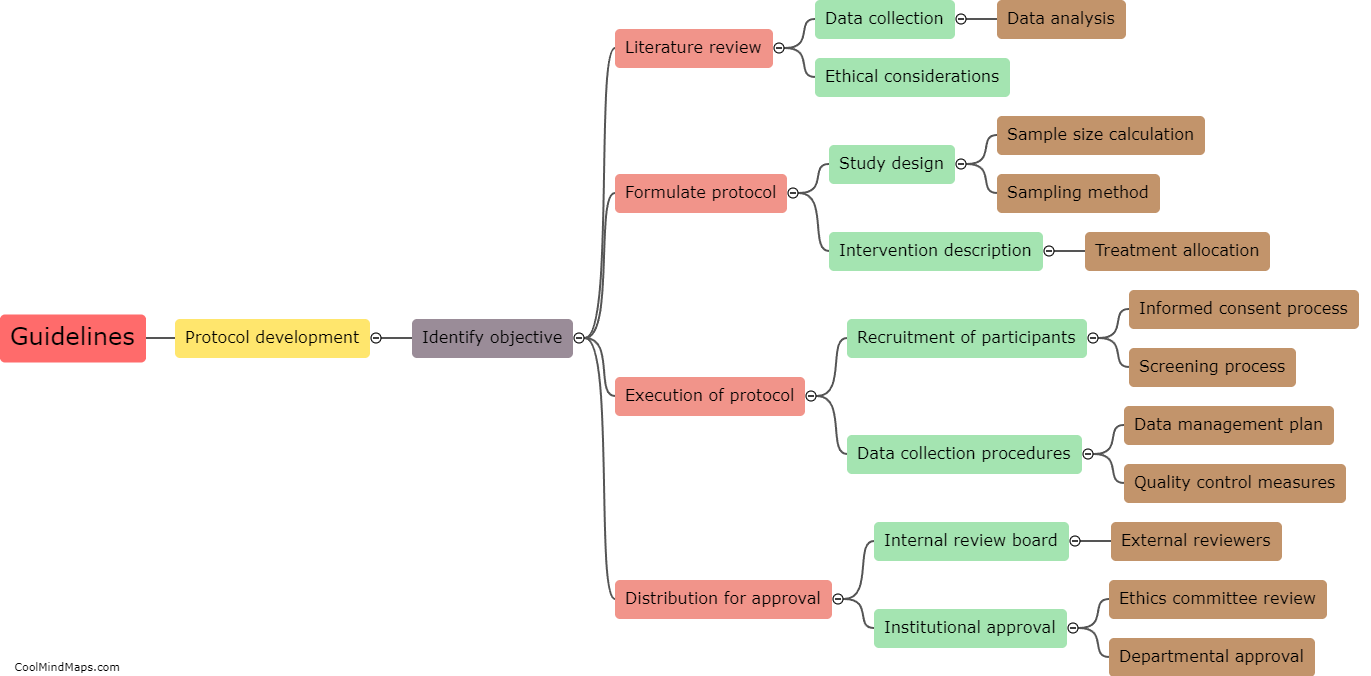
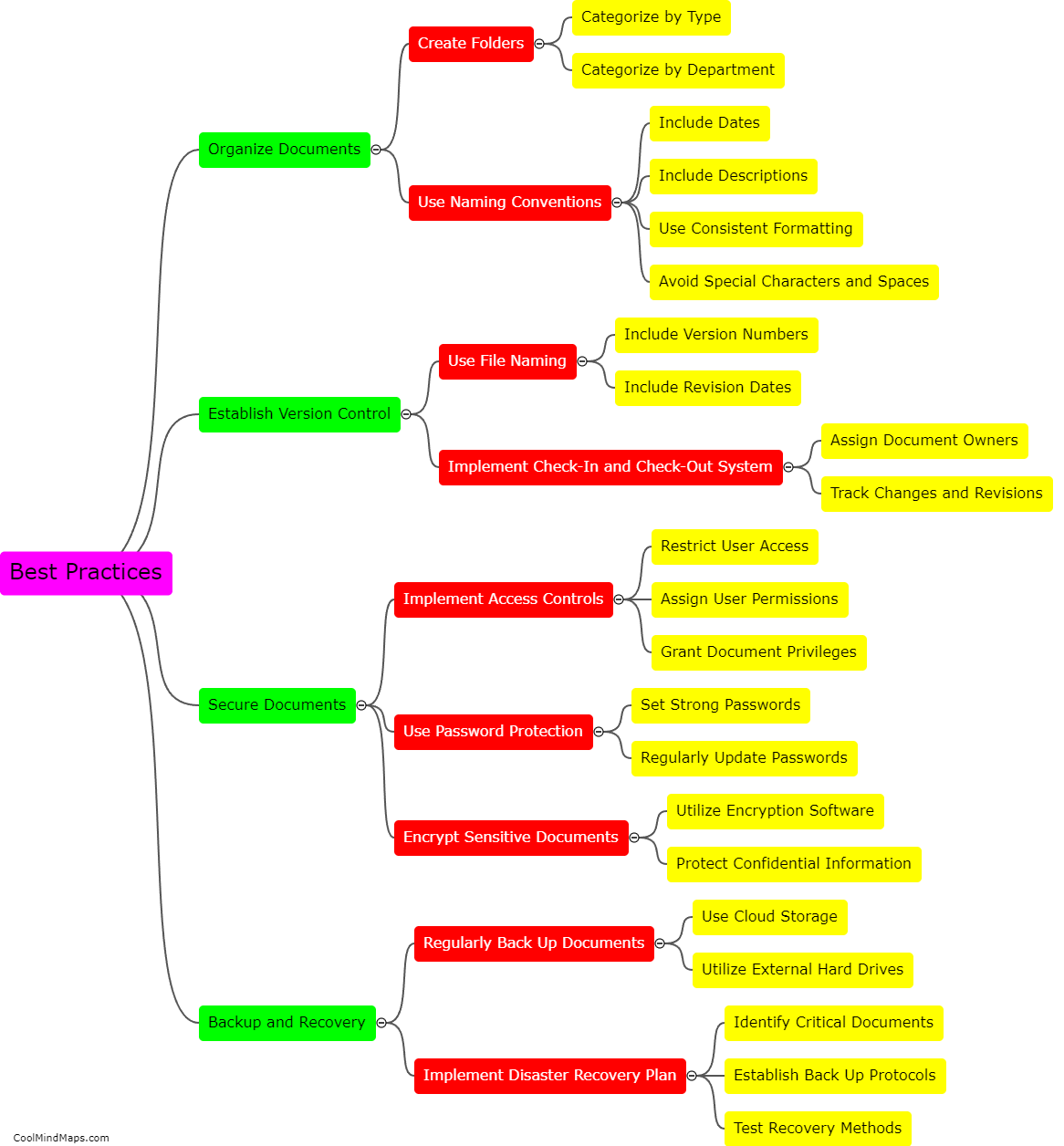
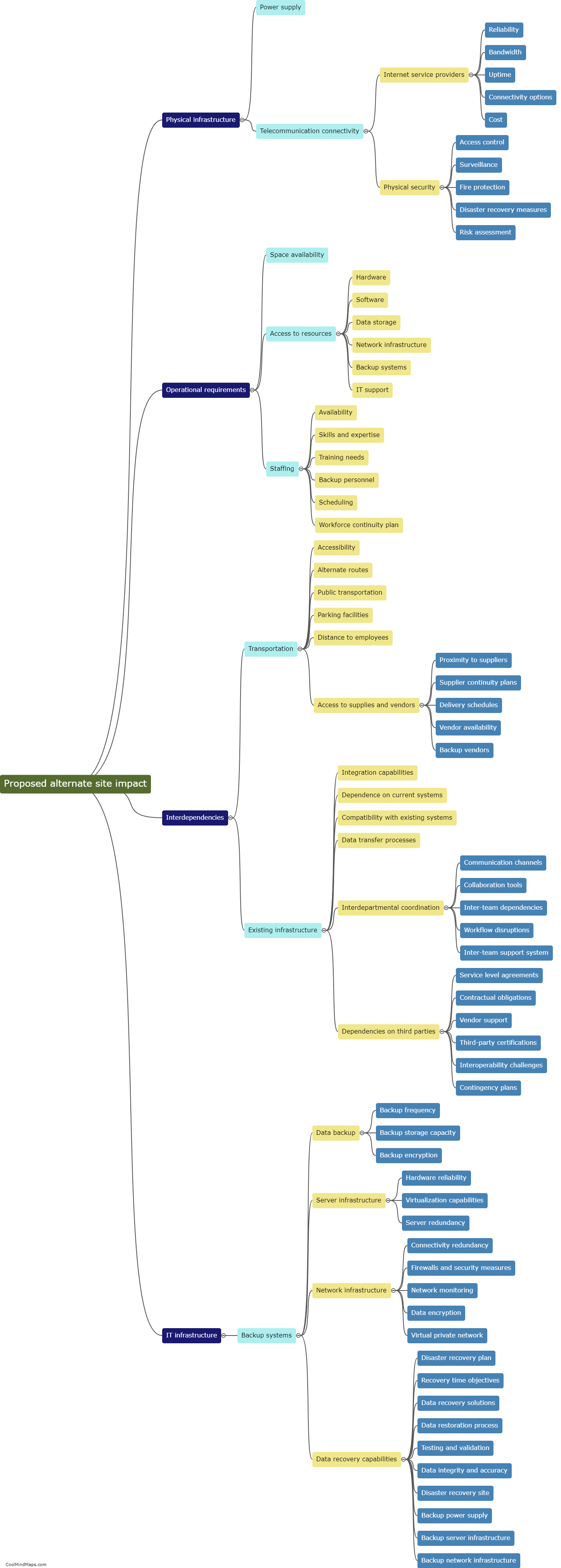
The regulatory requirements for essential documents in clinical research are an important aspect of ensuring the integrity and validity of research studies. These requirements are defined by regulatory authorities such as the Food and Drug Administration (FDA) and the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Essential documents refer to those documents that individually and collectively permit evaluation of the conduct of a study and the quality of the data produced. These documents include the protocol, investigator's brochure, informed consent forms, case report forms, and various administrative documents such as study logs and correspondence. Regulatory guidelines dictate that these documents must be complete, accurate, and maintained throughout the study, and they must be readily available for inspection by regulatory authorities. Compliance with these regulatory requirements is crucial for ensuring patient safety, data integrity, and the ethical conduct of clinical research.

This mind map was published on 5 December 2023 and has been viewed 96 times.